A new study led by Thomas Peterson Ph.D. ’16, biological sciences, reveals thousands of previously ignored, rare mutations that likely contribute to cancer. Peterson conducted the work as a student in the lab of Maricel Kann, associate professor of biological sciences, with additional contributions from Iris Gauran, Ph.D. student; Junyong Park, associate professor; and DoHwan Park, assistant professor, from the UMBC department of mathematics and statistics.
The research applied an innovative statistical analysis to genetic data from cancer patients. It focused on functional regions in proteins (called protein domains) within protein families that include proteins already implicated in cancer. Within this framework, even mutations that only appear in one or two patients could be considered meaningful if the mutations are in the same position in the protein domain as in other proteins in the same family.
The new method is “a really good way to identify important variants that you wouldn’t normally find in traditional methods,” says Peterson, “because we can look across gene families, which is something we normally don’t do.”
By identifying domains that are more likely to contribute to cancer, which the authors dub “oncodomains,” this study could help scientists prioritize particular areas within the very large field of cancer research. Their approach encourages drug development targeting the function of these domains, and because the domains are the same across so many proteins, it is possible that a single treatment could tackle cancers caused by a broad spectrum of mutated proteins.
It may even be possible to use the same statistical methods to help identify mutations that cause rare diseases. Many rare diseases may be caused by mutated proteins in the same families as those that cause more common diseases with more available data. Looking at protein domains across patients with several different diseases could point researchers toward the mutations that are most likely to be involved in causing a rare disease, even if there are only data from very few patients.
“Maybe only two patients have a mutation in a particular protein, but when you realize it is in exactly the same position within the domain as mutations in other proteins in other patients,” says Kann, “you realize it’s important to jointly investigate those different mutations.”
Peterson started in Kann’s lab as an undergraduate as a system administrator for her databases. Eventually, he shifted gears to independent research, and Kann found a project that tapped into his passion and curiosity. After publishing three papers as an undergraduate, Peterson decided to pursue graduate work in Kann’s lab and solidified his career path as a bioinformatician.
Now a postdoctoral fellow at the University of California, San Francisco, Peterson is expanding on the work he completed while at UMBC, and he remains connected to Kann’s lab by helping to train her new students to pick up where he left off.
“Tom made me realize how important it is for a mentor to find the project that best matches the student,” says Kann. “Nothing motivates a student more than working on a project he is passionate about.”
See the original article, Oncodomains: A Protein Domain-Centric Framework for Analyzing Rare Variants in Tumor Samples in PLOS Computational Biology.
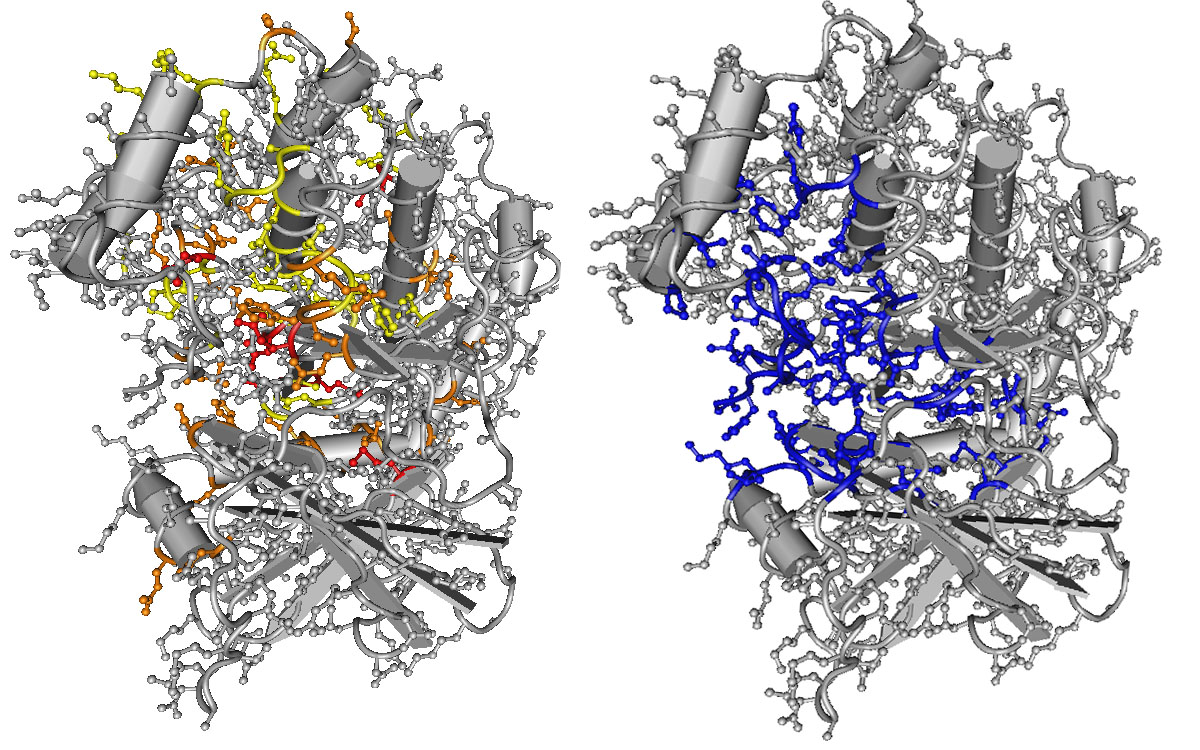
Image: Model of the structure of a kinase, a protein and enzyme. Positions that are most often mutated in disease (yellow, orange, and red, left) correspond with positions that are involved in the kinase’s primary function (blue, right). Figure 3 from Peterson et al. (2017).
Tags: Biology, CNMS, MathStat, Research


